Valence
Overview
Turning on the valence setting will enable the display of double bonds.
Toggling valence_mode alters the positioning of double bonds (for representation as Lines)
valence_size alters the distance of double bonds.
Note that bonds can be edited to be delocalized using Unbond and Bond.
There is also a command called valence.
Examples for the settings: valence and valence_mode
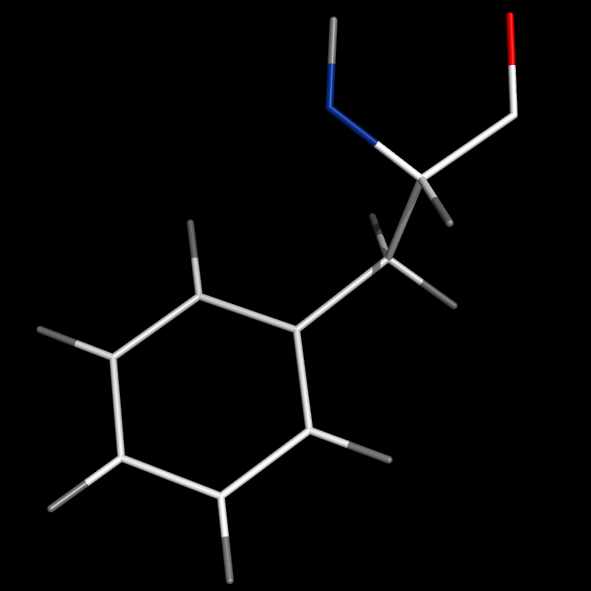
set valence, 0
#(no double bonds)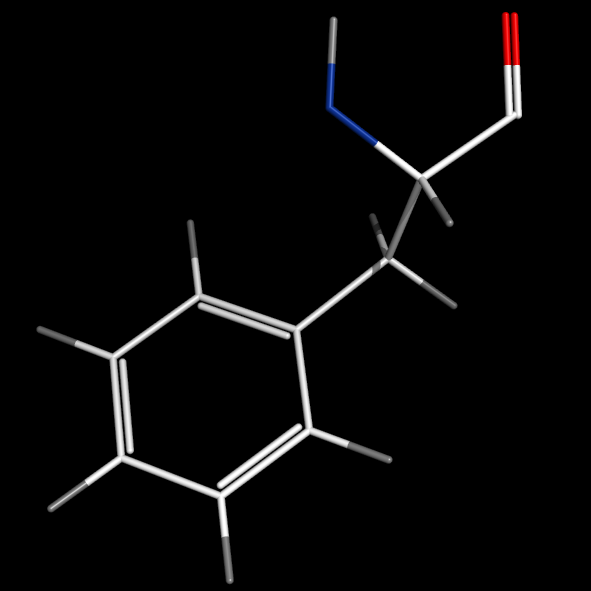
set valence, 1
set valence_mode, 1
#bonds inside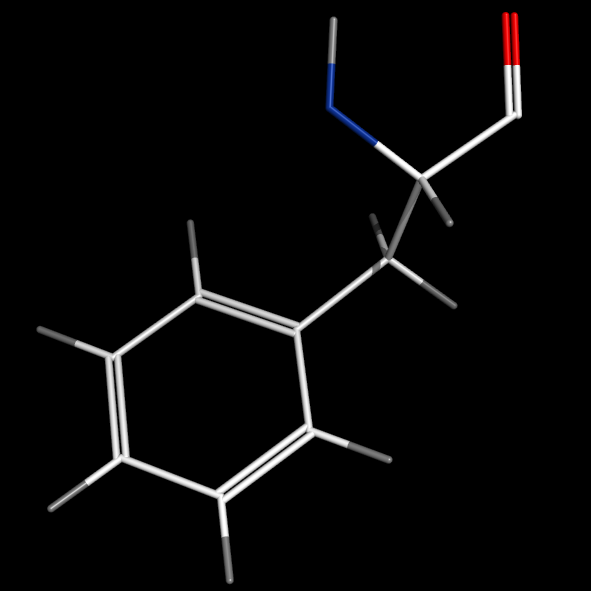
set valence, 1
set valence_mode, 0
#bonds centered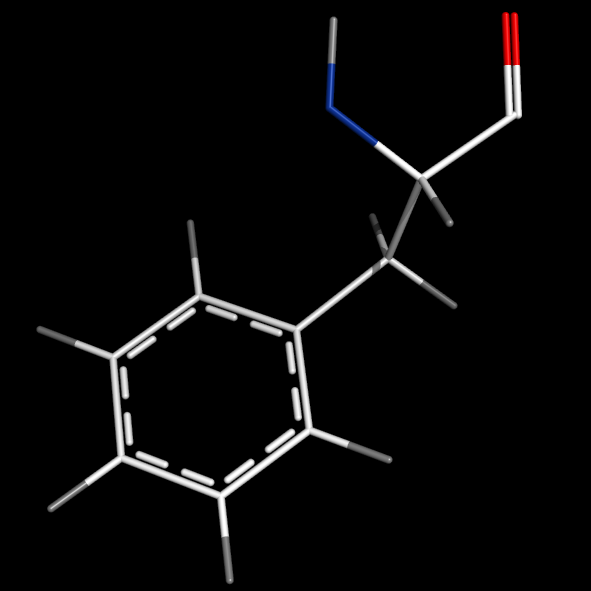
set valence, 1
#delocalized bonds
#see section "Editing bonds" below or try command "valence guess, all"




valence_size alters the distance of double bonds, but behaves slightly different depending on valence_mode
| valence_size with valence_mode 1 inside |
valence_size with valence_mode 0 centered |
|---|---|
 |

|
Syntax
set valence, 0 # off
set valence, 1 # on
set valence_mode, 0 # centered
set valence_mode, 1 # inside
set valence_size, 0.1 # default: 0.06 # range 0 - ~0.5
The valence command
The valence command automatically formats existing bonds and can even guess the bonds for standard amino acids.
#USAGE:
valence order, selection1 [, selection2 [, source [, target_state [, source_state [, reset [, quiet ]]]]]]
order can be either: 1, 2, 3, 4, aromatic, copy, guess
#make PyMOL guess/autoformat bonds in proteins
valence guess, all
Editing bonds
# In editing mode: select the bond using Ctrl-right-click, then enter:
unbond pk1,pk2
bond pk1,pk2,4
# 1: single bond, 2: double bond, 3:triple bond, 4:delocalized
Automatic editing of bonds
Try using the valence command first.
Secondly, Format_bonds is a script that automatically formats valence in all amino acids and has additional options.